WTFunding: Bioinformatics & Genetic Data

WTFunding is one of my “spare time” projects to delve into tech sectors attracting VC funding that pique my curiosity. I like connecting dots between disparate things, it’s also pretty useful.
Table of Contents:
- So what is bioinformatics?
- What are the applications?
- What’s hindering adoption?
- Who cares?
- What are the risks?
- What’s the current scene?
- Conclusion
So what is bioinformatics?
Simply put, bioinformatics is software used to understand biological data, including (but not limited to) genomic sequences. What’s particularly cool about the field is that it brings in a bunch of expertise and methods from other areas, like statistics and computer science in order to perform analysis and glean insights from this bio data.
If you only care about the business stuff and don’t really care about how it works (and / or hate science), then skip ahead to the “What are the Applications?” section.
How are living things made?
 The reference, for those who have had the misfortune of never watching “Jurassic Park.”
The reference, for those who have had the misfortune of never watching “Jurassic Park.”
Let’s start with what constitutes life. The simplest unit of a living thing is a cell; some living things just have one cell, whereas others, like humans, have something like 37.2 trillion cells. Each cell has a nucleus, which is home to most of the genetic material within cells. Within the nucleus lies chromosomes, which are made of protein and DNA.
At a lower level you can think of these cells as being made of molecules, which are atoms grouped together by chemical bonds. Cells depend on three macromolecules, or molecules with upwards of 1,000 atoms, to determine the cell’s’ basic functioning and structure: DNA, RNA and proteins. The relationship between them is that DNA makes RNA, and RNA makes proteins (this is the central dogma of molecular biology). For the computer nerds reading, think of DNA like persistent storage, RNA like volatile memory and proteins like executables.
Zooming further in, genes are regions of DNA that is the recipe book, so to speak, for biological function. They’re what gives a living thing its biological traits — for example, I have the “blue eye” gene. For a sense of scale, there are 23 pairs of chromosomes within the human body that include over 20,000 individual genes that are made up of over 3 billion DNA base pairs.
So what are base pairs? The building blocks of each strand within the famous double helix of DNA are called nucleotides. Each of these nucleotide building blocks is made of either guanine, cytosine, adenine or thymine — which are shortened to C, G, A or T. But, these building blocks pair off to form base pairs, and only like the same partners: G always goes with C and A always goes with T (G-C and A-T). So, if you know the blocks in one strand of the double helix, you also know the blocks in the other.
As I said before, DNA makes RNA and RNA makes proteins, but how? DNA contains exons, which get converted to messenger RNA (mRNA), which delivers the message of which proteins are needed in a cell. The official way to describe exons is as “coding” for proteins, since like a computer program, they end up telling the cell what proteins to have.
Introns, in contrast, don’t code for proteins. Their relevance to this discussion is that they happen to also show where individual genes are separated. That isn’t to say that they are useless otherwise — they’re quite important for regulating how proteins show up in cells, but that’s not important for what I’ll be covering.
This is a good illustration of how all these things link:
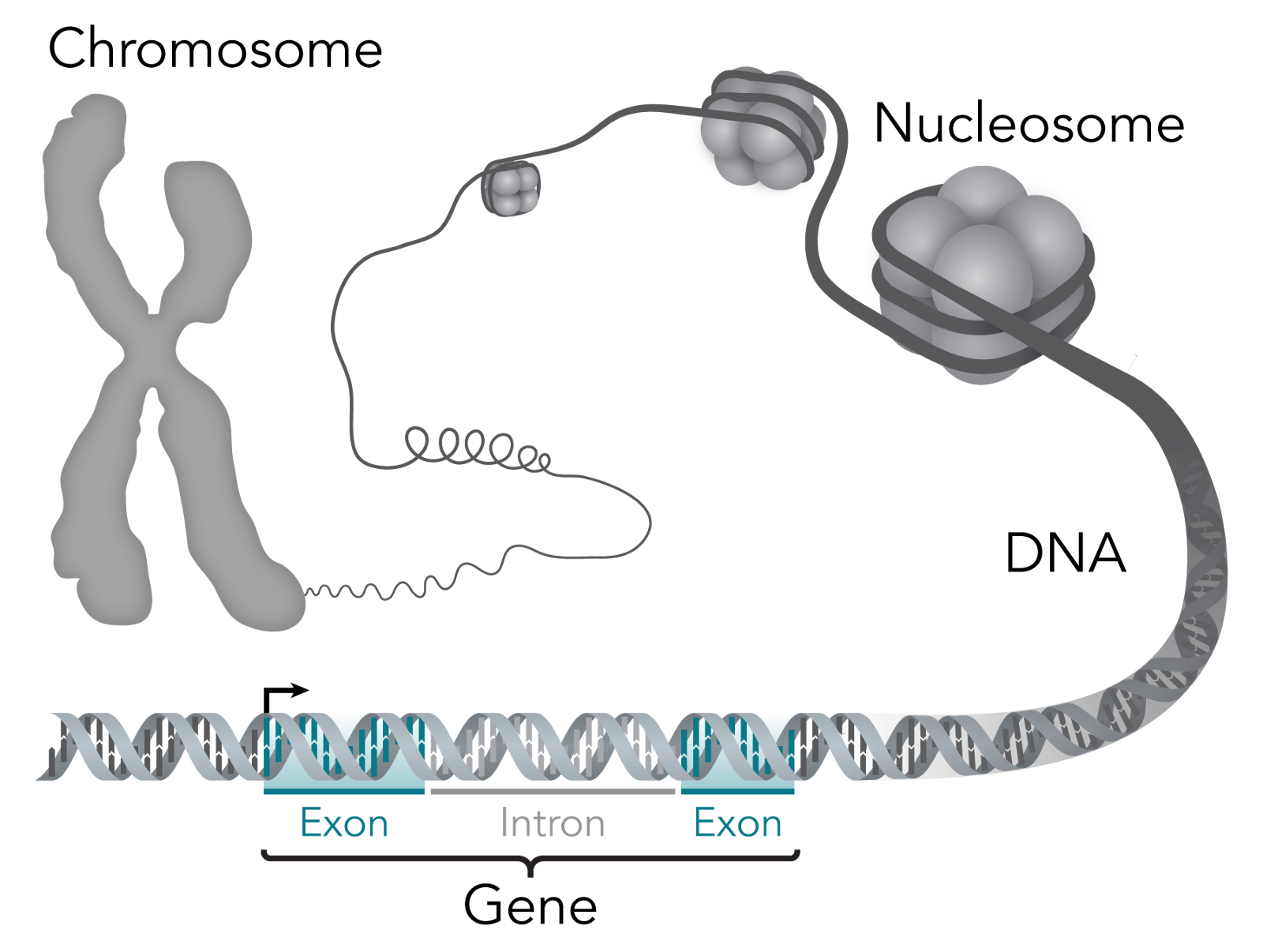
When you put all the genes within a living thing together, you get its genome. You might be familiar with the Human Genome Project, which is an international research body launched in 1990 dedicated towards mapping out the “human genome.” I have “human genome” in quotes because what they mapped out is an amalgamation of genomes — each human being has a unique genome with special variants in genes. They accomplished their goal in 2003, making it a warm and fuzzy example of the world banding together towards the common good.
The project also spurred a lot of innovation in the field. The way they were going about mapping the human genome was through DNA sequencing, the method for which at the time hadn’t evolved since the 1970s, other than getting a boost as computers became more powerful. But, concurrent with the start of the Human Genome Project, new sequencing methods, novelly called “next generation sequencing” methods, or NGS for short, began being developed and had gained a market foothold by the time the project finished. So, what exactly does DNA sequencing entail?
DNA Sequencing Process

Before you can figure out the purpose of specific genes, the DNA in question must first be sequenced. For simplicity’s sake I’m sticking with DNA sequencing in this section vs. other types of sequencing, because an already lengthy post would have to become a novel.
DNA sequencing just means determining the exact order of the building blocks (nucleotides) within the DNA, and more specifically the order of its bases — the Gs, Cs, As and Ts. I’ll walk you through a next-generation sequencing (NGS) process, since that’s been the catalyst for rapidly decreasing costs of sequencing, although there’s also the Sanger method, which is the “old school” method.
Library Preparation
1. DNA Samples
First you have to start with some sort of sample from which to extract DNA. It can be from blood, fossils, saliva or tissues, although saliva is now the go-to for collecting samples from humans given its ease of procurement.
2. Starting Material
But, there’s also a lot of other stuff in saliva (or any of the samples) that needs to be weeded out. Isolating DNA is done by “purifying” it from a sample through lysis, which disrupts the cell in order to release and separate its biological contents. There are physical and chemical methods for performing cell lysis, the coolest of which is probably sonication — harnessing ultrasonic sound energy to fragment the cell.
Once the cell is broken open, the non-DNA contents are removed via chemical methods, like adding detergents to remove the cell’s surface materials and enzymes that break down proteins and RNA. But those chemicals also have to be separated, so some extra chemistry magic happens (or a wash / spin cycle in a centrifuge) to get the DNA to bind together. Now the purified DNA can be extracted as starting material. As with many things in life, the more starting material, the better.
3. Fragmentation
Now, if we’re talking about human DNA, the genome is going to be really long and would thus take a really long time to sequence. So, the next step is cutting the DNA into smaller pieces for sequencing to help speed up the process. These fragments are officially termed “reads,” which I’ll use going forward. And fittingly, the collection of DNA fragments you generate is called a “library.”
But how do you determine how big you want these fragments to be? It largely depends on how the fragments will be sequenced and for what they’re being sequenced. Most NGS methods sequence up to 400 base pairs (“bp”) during one sequence cycle (called a “run”); the old-school Sanger method typically sequences up to 900bp.
The two typical approaches for this are either physical or enzymatic. One example physical method is acoustic shearing, which may be even cooler than sonication; the DNA sample is placed into a glass vial which is then subjected to acoustic energy that continually creates and collapses microbubbles. The process of growing these bubbles and causing them to implode creates shockwaves that have sufficient power to break down the sample DNA into random fragments. The power of the microbubbles fragments DNA pretty quickly with little loss of the DNA sample, and creates fragments ranging from 100bp to 5,000bp.
The other common physical method is nebulization, involving nebulizer devices which use compressed air to convert liquids into a fine mist. DNA is pushed through a small hole in the nebulizer, creating a mist which is then collected. The resulting fragments are typically 500bp to 3000bp, depending on how quickly the DNA is forced through the hole. This method is pretty quick, but can cause DNA to be lost in the process.
Enzymes capable of degrading DNA can also be used to fragment it into smaller pieces. Although it’s more consistent than physical methods, it also has the opportunity to alter the fragment by insertion or deletion. So, your method of choice really depends on your end goal.
4. Repairs & Adapters
First, let’s take a look at how a DNA molecule looks, broken apart by its ribbons (or strands):
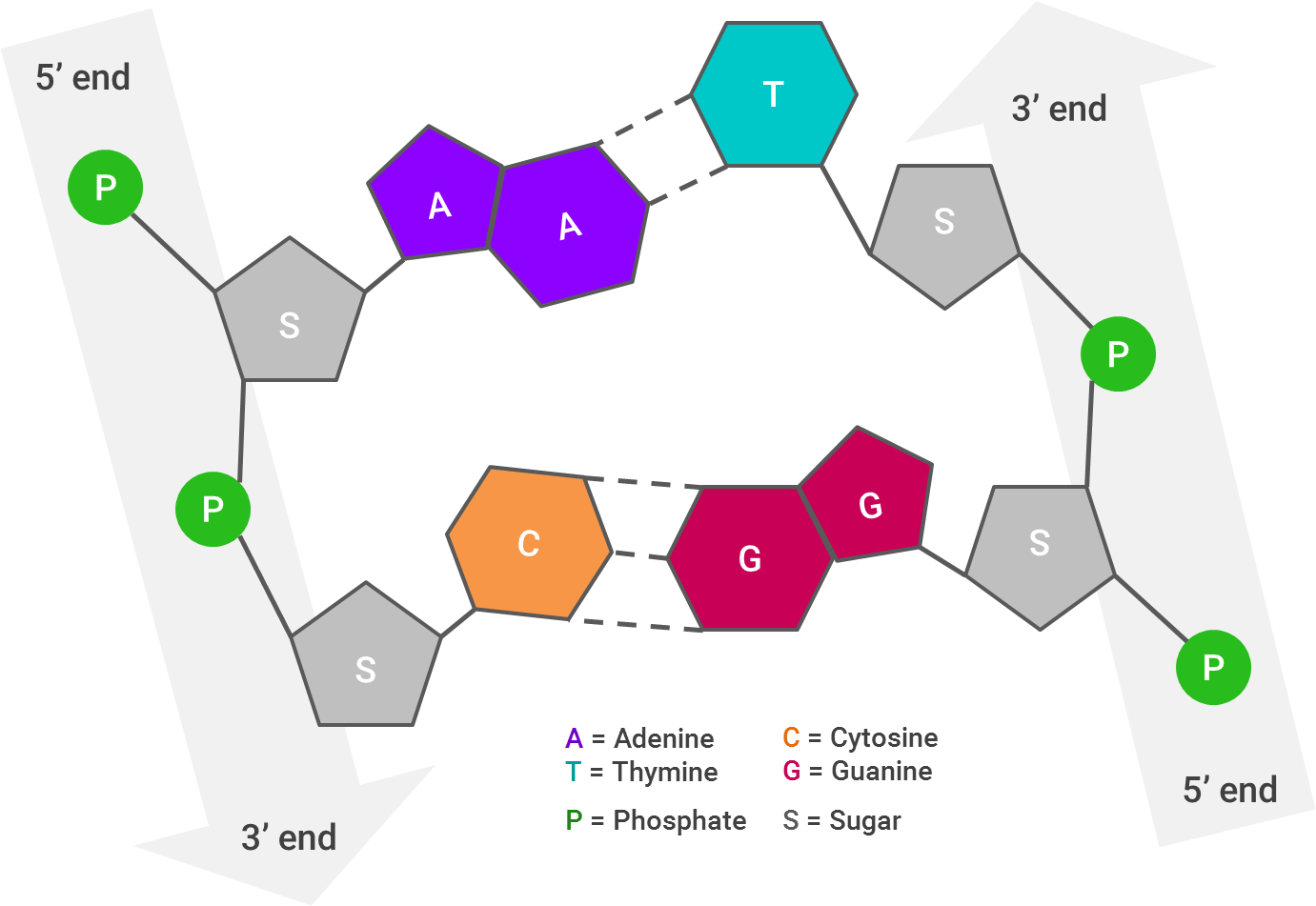
Each arrow represents one strand of DNA; I’ve laid this out linearly, but as you very likely know (and if the abundance of pictures in this post hasn’t reinforced it), the strands twist to form a double helix. The 3’ end of one of the DNA strands aligns with the 5’ end of its partner strand.The way the DNA molecule is “read” is from 5’ to 5’. So, if the 5’ ends of both strands stick out farther than each other, the strands will be “repaired” by combining them together and filling in the gaps (A with T and C with G).
This is crucial for the next step, which is putting “adapters” onto the DNA strands. Adapters are short DNA molecules that have been synthesized specifically to help in the sequencing process. Adapters are generally provided in a kit, and the adapter sequences therein will be added to the 5’ and 3’ ends of every fragment within one library.
Some may help put “primers” into their correct place, while others may help the DNA fragment stick to a surface in what’s called “immobilization.” The primers have to match the beginning and the end of the DNA fragment, since they’ll serve as the guide for what DNA fragment is amplified in the next step. The final step in the library preparation, immobilization, just means that each single DNA molecule is made up by a bead, which is then anchored to some solid surface, like a glass plate.
Amplification
Polymerase chain reactions (PCR) is used effectively as a copy machine for a DNA sequence, or what’s known as “amplification.” This thinking is actually somewhat similar to the methods for improving image quality within satellite imagery that I discussed in my prior post. If you think of the target DNA as an object to be captured within a landscape, it’s far easier to take a bunch of quick pictures of it than take one big, detailed picture of the landscape and try to extract the object from it.
This process follows an exponential curve, too, since you’re making copies of the copies. After just 6 cycles, you have 64 copies of the target gene. After ten, you have 1,024. These copies then make up a “DNA colony” to be sequenced.
Sequence Reaction
 Note: this is a sequencing UI from a TV show and thus should be disregarded entirely as resembling reality
Note: this is a sequencing UI from a TV show and thus should be disregarded entirely as resembling reality
NGS is also known as high-throughput sequencing, which just means that there’s tons more data that comes out of the sequencing process, and it’s due to parallelization. There are a few different sub-methods of NGS, the most common of which seems to be Illumina’s sequencing by synthesis (SBS) — though the others also depend on the sort of library preparation and amplification discussed above.
In SBS, the immobilized DNA is “washed” with one of the four nucleotide bases (either T, A, C or G) at a time to see which gets incorporated. For example, if the fragment’s template is GCGAATCG, and your wash is “A”, both of the A’s in the middle will incorporate the wash and the others will be washed away. Then, using the power of lasers and fluorescence, a machine can record which part of the fragment incorporated the base, showing _ _ _ A A _ _ _.Then, the “A” dye would be chemically removed so the next cycle can start, a “G” wash would be used, the laser magic happens, and the machine would record G _ G A A _ _ G, and so forth one nucleotide at a time until the sequence is complete.
For frame of reference, these machines are really, really expensive. At the low end, Illumina’s Miniseq “benchtop” sequencer is about $50,000 and produces an output of up to 7.5GB in just 24 hours. But at the high end, its heavy-duty sequencer, the HiSeqX, is sold for $10 million and only in units of 10, producing an output of up to 1.8TB in less than three days. Even despite their hefty price-tag, it’s still far cheaper and faster than Sanger sequencing (the “old school” method).
Sequence Assembly
The process above generates sequencing “reads,” but not the genome itself — which is where sequence assembly comes in, and is also where data science starts to get involved. De novo sequence assembly, which seems to be the most commonly used, reconstructs an organism’s likely genome sequence based on the reads. The word “likely” here is important, since there’s no guarantee that it’s correct, but instead is approximating the source of these fragments.
As you might guess, this leaves some room for error, which I’ll touch on in the “what’s hindering adoption” section. Errors can be in the form of false negatives, such as thinking a fragment is a mistake or just a repeat, or the fragments could just be linked up together in the wrong spots…plus there’s no guarantee that there weren’t errors in the sequencing process itself. To better illustrate why this process is so tricky, Wikipedia has a great analogy:
The problem of sequence assembly can be compared to taking many copies of a book, passing each of them through a shredder with a different cutter, and piecing the text of the book back together just by looking at the shredded pieces. Besides the obvious difficulty of this task, there are some extra practical issues: the original may have many repeated paragraphs, and some shreds may be modified during shredding to have typos. Excerpts from another book may also be added in, and some shreds may be completely unrecognizable.
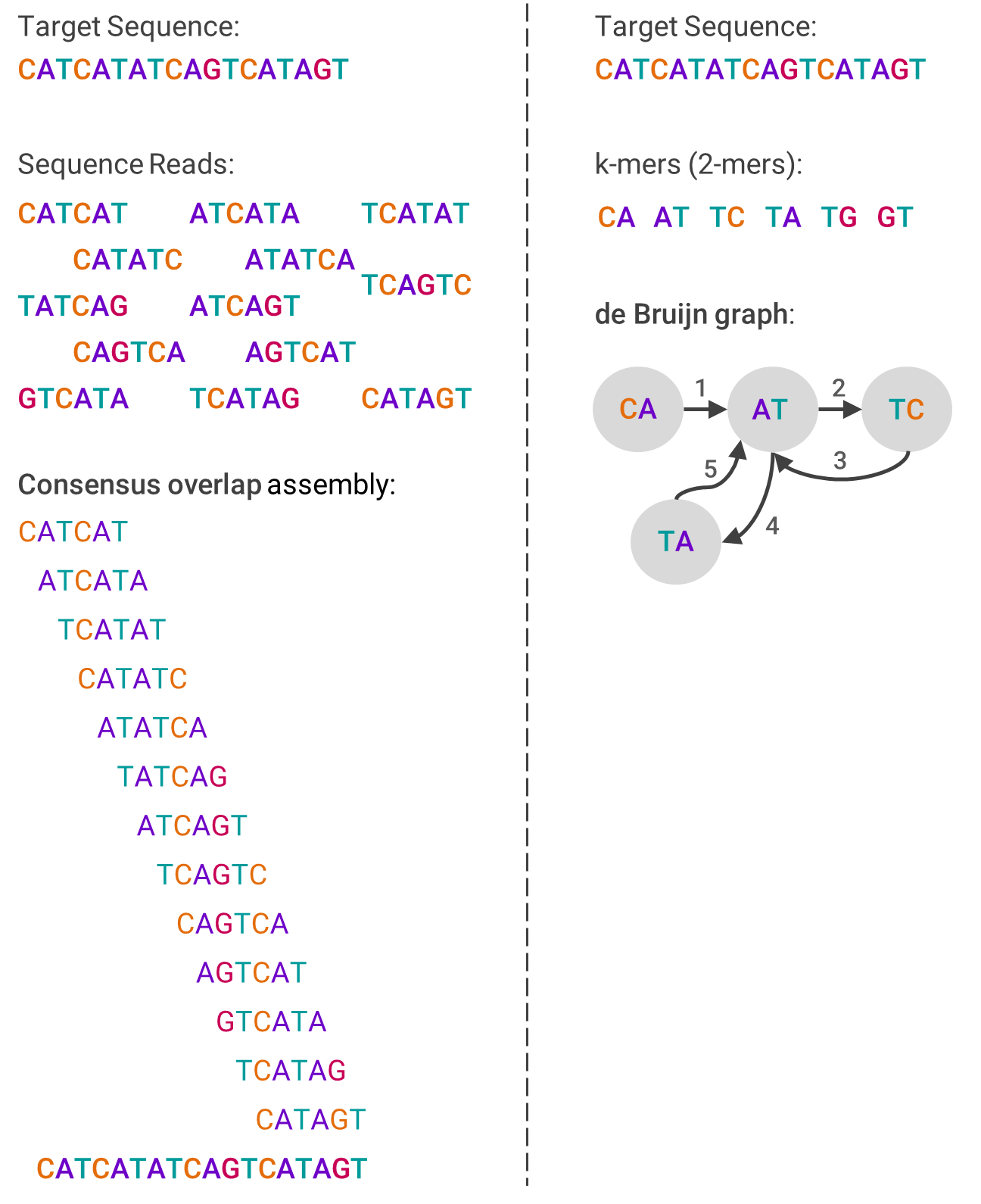
One example algorithm used in de novo assembly is overlap-layout-consensus (OLC), which leverages a graph to represent the reads. First, overlaps between fragments are computed, and then each fragment is connected by an edge if there are indeed overlaps. The computing problem then becomes how to navigate through the graph in a way that still contains each fragment, which involves a lot of graph theory.
The other primary assembly algorithm is the de Bruijn graph (DBG), which also is based on graph theory. First, the reads are broken into smaller pieces of the same size (called k-mers). Then, the graph is created by ordering from the pieces that overlap at the beginning of the sequence to those that overlap at the end. This sounds linear, but isn’t.
The image below should help visualize each algorithm and compare how they differ:
To move on, let’s assume for now you’ve done a pretty good job sequencing and assembling the DNA in question. It’s not just there to be admired, but to gain something useful out of it, which leads into sequence analysis.
 An example of a DNA sequence and foreshadowing of my “what are the risks” section
An example of a DNA sequence and foreshadowing of my “what are the risks” section
Sequence Analysis
Sequence analysis is where the startup software solutions addressing bioinformatics begin to appear. This is the process of taking the sequenced DNA and applying meaning to it — determining and reporting what the genes are and what is their purpose.
Homology Search
“Homology searching” is a fancy way of saying looking for similarity between sequences, and is step one (at least to try) towards getting to the end goal of understanding the genes you’ve just sequenced. “Homology” itself means shared ancestry between genes in different species — after all, all species evolved from the same ancestors. So, when sequences between two distinct species are similar, you can argue that there’s homology.
If you are able to search a database of known sequences to see where they might be similar to your sequence, then you can much more easily identify certain genes and their functions, as well as speculate on evolutionary relationships. In an ideal scenario, if the results of your homology searches cover all the genes you need identified, and they already describe what the genes are for, then you’ve arrived at your end-result. You’ve also saved yourself a lot of other potential steps, which are described right after this.
So how do researchers compare sequences? It involves a lot of data science and also necessitates having a rigorous dataset with which to compare. The more genes are identified on other species, such as mice, the greater ability there is to search human DNA sequences for similar genes.
The most commonly used tool for homology search is BLAST, for “Basic Local Alignment Search Tool,” which is favored due to its speed, at the expense of some accuracy. For those familiar with statistics, it’s a heuristic algorithm. The way it compares a query sequence to a sequence database is by finding “high-scoring pairs,” or HSPs for short, which represent overlaps that seem statistically significant. The speed comes from creating “words” within a sequence, which are generally 11 letters long in DNA sequences, and running the search on each word, narrowing down which HSPs fit as the search moves through the letters. This constitutes a substitution matrix, and would look something like this in BLAST:
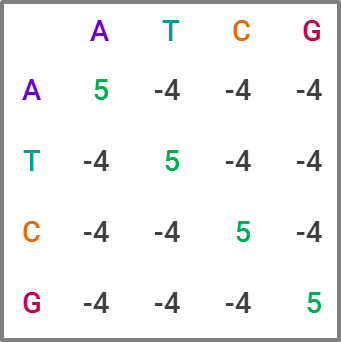
While BLAST is the current market leader, improving upon it is clearly an area of interest within academia. Like in many data science projects, making an algorithm both faster and more accurate is the holy grail. For projects with lots of sequence data, BLAST becomes computationally expensive — and given the explosion of sequence data in recent years, researchers have developed and proposed alternatives that potentially offer greater efficiency and ameliorate computational cost concerns. I could publish an academic journal on all the approaches proposed, but I’ll explain one I find particularly neat and badassly-named, GHOSTX.
GHOSTX’s speed advantage is by analyzing both length and match score — the algorithm will only ingest sequences that are both close to the appropriate length of the target sequence and deemed to be statistically similar content-wise. To oversimplify a bit, it’s able to do this by using suffix arrays, an example of which is below:
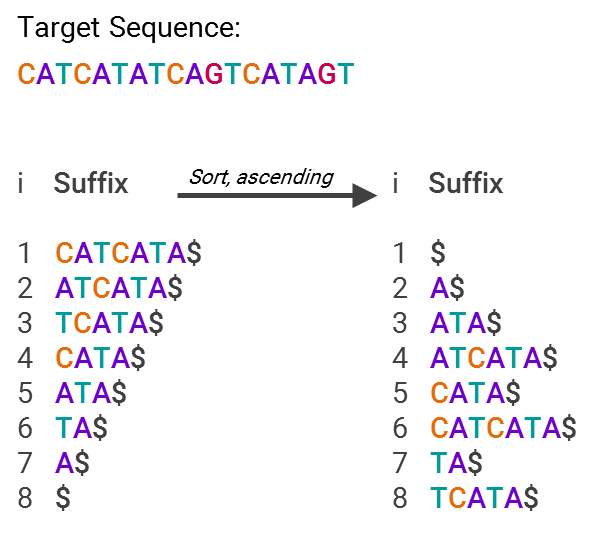
This is much more efficient for searching, since now you just have an array with pointers to text snippets (suffixes) that are sorted in alphabetical order. So, what’s being stored isn’t “A$,” “ATA$” and so forth, but “7” or “5”, that is, what’s stored is only the code for where in the list the suffix lies. In other words, you now have an index for all the different suffixes, making it easy to find where your sequence matches others in the database.
Using these various methods, even if you find a matching result for a gene, there still may be insufficient data on what the gene does (i.e. lacking gene annotation, which I’ll describe in a bit). In those cases, and others in which homology search doesn’t complete the puzzle, you’ll have to pursue the following steps in addition.
Gene Identification
If the homology search fails, or for whatever reason you just don’t want to perform a homology search, gene identification is the next step. What you’ll be looking for is the region of DNA that specifically codes for its function, i.e. codes for protein. These are called open reading frames (ORF), which you can think of as triplets of base nucleotides (again: A, T, C or G), and researchers use them to initially identify regions that probably show what the gene is.
Fancy data science can be involved in this step, too, as gene prediction (which is just the predictive form of gene identification). There are three primary methods of doing so, which include statistical, comparative or empirical methods (i.e. homology, which I went through above).
As an example of a statistical method, gene prediction programs using neural networks can be trained on the organism whose DNA is in question and then applied to the target sequence. Hidden Markov Models (HMMs) also use training data to return the most likely structure of a gene, which is useful if there’s no gene in a sequence or only a partial gene.
Comparative methods use DNA from another species to help predict gene function, since there’s a lot of overlap in how our genes work. The common example is using mouse DNA to determine genes in human DNA; after all, we’re a lot more like a mouse than we are a plant. In fact, 95% of genes that actually code for a biological function match one for one between mice and humans. The theory behind this is that evolutionary pressures led to a general, optimized blueprint for how mammals should function.
However, both these methods still require experimentation to verify that the gene prediction is accurate…at least for now, until these methods (particularly statistical) become more accurate.
Translation & Database Search
Once you have identified your gene, whether through homology search or gene identification, you need to translate that gene into its proteins — the stuff that brings to life whatever the genes have blueprinted. These translators are pretty easily found on various academic websites. For example, the sequence I’ve been using in my examples throughout, “CATCATATCAGTCATAGT,” by pure luck happens to have an ORF (the region that actually codes for proteins). It’s in “ATGACTGATATGATG,” which is in the “opposite” DNA strand of my sequence (specifically, the opposite of “CATCATATCAGTCATAGT”).
Once you have your ORFs, you can search them against protein databases to see if there’s already data on them. If there is, it’ll help enormously in the next and crucial step, gene annotation, since you’ll have a head start on knowing what purpose the gene serves.
Gene Annotation
Gene annotation is the process in which a specific gene is assigned a specific functional or physical feature. This ideally is largely automated and just enhanced with human expertise, because it would take a very long time for one person to manually annotate a DNA sequence.The functional features generally are drawn from a specific vocabulary to form an ontology, or a formal method of naming and defining things.
There are two components of annotation: the gene’s structure and its function. Structure mostly just determines which regions code for proteins, while functional annotation records the gene’s biological and biochemical functions. Its function just means what proteins are there, or what proteins are hypothetically there (such as predicted by gene finding programs), which engenders how the gene influences an organism’s operations. The gene is also given a name, whether unknown or known based on being identical to a previously documented gene.
Automated annotation uses data science in order to help predict the relevant gene annotations, like its functions. Bayesian networks and similar statistical methods are used by learning from prior annotations, but doesn’t help with finding new gene functions. Automated annotation certainly helps speed up the process, but it’s basically impossible to be accurate on the whole genome, so manual annotation is still needed to clean up any errors or fill in the gaps.
Annotation is one of those things that sounds really easy to do (“just write down what the gene does!”), but is actually really tricky in process. The quality of annotation is largely dependent on the quality of the process before it. If the gene prediction was faulty, or if the sequencing shoddy, it will result in greater difficulty and / or error in annotation.
Upload & Stop, Collaborate and Listen
 This is not the sort of DNA data storage I mean.
This is not the sort of DNA data storage I mean.
Once the DNA has been sequenced and annotated, it has to be stored…which sounds simple enough, but isn’t quite. In a brief crossover to my industry, security and compliance is actually pretty important when considering biological storage; after all, someone’s genome is extremely private information (though admittedly a mouse probably doesn’t care if their genomic data gets hacked).
Adding to the complexity of storage, there are various types of databases for genomic data — some that contain empirical genomic data, predicted genomic data or structural data. It might also have just the raw sequencing data, or data that’s been cleaned up a bit. Some are publicly available, but some aren’t. The databases also have to be able to facilitate search across sequences, too.
While databases may be a bit passé in other areas of tech, this is actually a reasonably hot area within bioinformatics. Making it easy for customers to search and combine different types of genomic data, such as via an API, is hugely useful in bioinformatics applications, but even helping them manage their data is a plus as well, such as through versioning control (since this sort of data is always being updated). Some startups are also focused on unifying this data with other sorts of medical data, such as patient outcomes, which helps streamline their customer’s processes considerably by not requiring them to visit lots of different databases and tools.
Collaboration is another key part of the bioinformatics end-stage. For example, when designing a pharmaceutical drug, the R&D team needs to collaborate with the clinical trials team, who needs to collaborate with the clinical team in order to maintain a complete feedback loop. So, bioinformatics software is also about optimizing workflows to produce more robust results — which may sound ridiculously outmoded to techies at the cutting edge, but, well, there it is.
 The O.G. thought leader.
The O.G. thought leader.
Now with these query-able databases of annotated genes, i.e. you have your sequenced DNA handy and know more or less what the random letters mean in a biological sense, you can now get to work on using them towards a variety of use cases.
What are the applications?
While genes really shouldn’t be thought of based on what diseases they cause, the most obvious applications of bioinformatics revolve around discerning better treatments of diseases. The ability to map out genes and relevant mutations of complex and pervasive diseases such as cancer, diabetes or those that cause infertility could help research in which drugs or other forms of therapy might alleviate — or even eliminate — them.
With that lens, there are three major areas that can benefit the most from bioinformatics: pharmaceuticals, personalized medicine and genetic testing.
Pharmaceuticals

Pharmaceutical companies can leverage bioinformatics for a variety of applications, from target discovery and drug design to improving the efficacy of existing drugs.
Pharmacogenomics studies how people’s responses to drugs are affected by their genes towards a more tailored approach to pharmaceuticals. You also might hear the term “pharmacogenetics,” which is typically used interchangeably with pharmacogenomics but does have a slight difference. Pharmacogenomics involves looking for differences between people at the genetic level that can explain drug response, while pharmacogenetics involves looking for a genetic reason for why there is a specific drug response. So think of pharmacogenomics as a whole-genome approach while pharmacogenetics deals with just one interaction between a drug and genes.
Right now most medications take a “one size fits all” approach, when in reality a person’s genetic makeup may determine how beneficial a drug is or what side effects they experience. In the future, this means doctors could analyze your genome against a variety of drugs for a specific condition to optimize your therapy — part of personalized medicine, which I’ll discuss in greater detail below.
This type of “predictive” approach would be a big benefit to patients, as they would theoretically no longer have to suffer through a trial and error period that can sometimes come with harmful reactions. Everything from the type of drugs to the dose amount could be tailored based on your predicted reaction to the medication. And, when the data on your response based on this predicted approach is recorded, it would help improve prescription algorithms, resulting in a huge data-network-effect.
For example, at least one in ten Americans takes antidepressants. Which antidepressant and dosage works for some people vs. others currently is more art than science. Further, these medications can often come with adverse side effects that reduce the patient’s quality of life — from insomnia to emotional numbness — or even make their depression worse. If instead their genome could be used to accurately predict which drug and how much of it would improve their depression the most, or at least cause them to suffer the least, their outcomes would be far more quickly and painlessly reached.
So how would pharmaceutical companies reach this stage? Genetic data could be used to isolate a particular protein related to a disease and conduct research towards finding a drug effective against this protein. Or, they could use genetic data to better understand receptors (such as a protein) so they know which would be the best target for their drug molecule. There’s also plenty of room for improvement with existing drugs; oftentimes drugs are effective against conditions for reasons that aren’t well understood, and bioinformatics could help determine why that is.
Ultimately, the real dream for the future of pharmaceuticals is a combination of bioinformatics-informed drug development with a feedback loop of patient outcomes to continually inform and improve drug design. This would likely serve as the cornerstone of personalized medicine, which leads us to…
Precision / Personalized Medicine
 In 2030, scientists will discover the key to all health issues is giving someone a puppy.
In 2030, scientists will discover the key to all health issues is giving someone a puppy.
“Personalized” or “precision” medicine is all about tailoring treatment and prevention based on an individual’s unique characteristics, including their genes. It’s unrealistic (or so my 2016 brain believes) to assume that in 2100 AD there will be a unique drug for each person and their conditions, but it is realistic to assume, even in the nearish future, that populations can be segmented in a way to substantially improve treatment. This isn’t just about drugs, either — it extends to medical devices along with prevention of diseases, too.
Part of this nearish future is improving the clinical trials process, the experiments performed with human participants to test the efficacy and safety of treatment methods. As discussed in the pharmaceutical example above, having a record of genetic compositions of potential trial participants can help improve the decision-making behind who should be included in the trial. With the clinical trials market estimated to reach $72 billion by 2020, you can see why having the ability to more carefully select trial participants in order to optimize clinical trial outcomes is worthwhile.
In a somewhat extreme case, Estonia is using a government-driven approach for furthering along these initiatives — it’s collecting DNA of all its citizens to create a genetic biobank. While right now the use case is primarily genomic research, farther down the line, having a strong sense of the average genetic “posture,” so to speak, of the Estonian population could help inform public health policy and population-wide personalized medicine.
Here in the United States, Obama created the Precision Medicine Initiative (PMI) in January 2015 within the Health and Human Services (HHS) department, primarily led by the National Institutes of Health (NIH). Its goal is simply to do away with the aforementioned “one size fits all” approach to improve medical outcomes for patients. It’s still in its early innings and has a somewhat slow timeline; in February of this year, NIH announced that it will study the genomes of one million volunteers by 2019.
The hope is the PMI will help kickstart precision medicine both by collecting a large swath of data as well as through developing improved analysis capabilities. It’s likely that everyday citizens won’t see the fruits of personalized medicine until quite a bit after 2019, as personalized treatments will require research, development and testing time, even once the genetic data collection and analysis methods improve. The presumable safety regulations around personalized medicine also may cause snags and delays, which I’ll discuss a bit more in the next section.
Genetic Testing
Genetic testing arguably must serve as the basis for any personalized medicine initiative, as only by knowing an individual’s genes can any treatment plans be tailored. A genetic test is simply a test that identifies either the person’s genes or changes in their genes (including proteins). There are currently a few uses for genetic tests, or at least uses cases leveraged in marketing.
Parents or couples considering becoming parents can use genetic testing to evaluate what sort of genes they will pass along to their offspring, generally with the goal of identifying genetic disorders. For couples pursuing in vitro fertilization (IVF), genetic testing can also find defects within the embryos created via IVF before they are implanted into the carrier, which increases the the embryo’s viability.
Individuals can test their own susceptibility to disorders and see what sort of mutated genes they have, or use it as a diagnostic test if they have a currently unidentified ailment. Or, more humorously, as I heard from a friend, being able to tell a sibling that you have less Neanderthal DNA than they have. This, in theory, helps individuals take preventative actions to stave off diseases or illnesses later in life.
While most genetic tests are currently based on DNA, longer-term there will likely be tests involving RNA and proteins. As discussed in the first section, DNA is the blueprint; this means it’s potentially a decent predictor of genetic risks, but not great at measuring the body’s current state for diagnostic purposes.
Genome Editing
While this isn’t a major application (yet), it’s pretty cool and a company doing it (Intellia) just IPO’d, so I want to touch on it briefly. Turns out you can also leverage DNA sequences and knowledge of genes’ functions in order to modify an organism’s DNA sequence, such as a virus.
The key tech enabler here has been CRISPR, or “clustered regularly interspaced short palindromic repeats,” which actually is part of a bacterial immune system. To use a reverse analogy from my industry, it’s a bit like most anti-virus software, where “signatures” of viruses are kept around so the system knows how to defend against it in the future.
The next part is using CRISPR-associated proteins, or “Cas,” that will actually chop virus DNA in half like a pair of scissors upon confrontation, the result being that the virus can no longer replicate.
This is pretty cool stuff, and while most of the research leveraging this technology is towards curing diseases, this could make the “designer baby” fears a reality, or even facilitate genetically engineered bioweapons (more on the latter in the “risks” section).
What’s hindering adoption?
 In the future, apparently we’ll be featureless and wear latex suits with our genomes printed on them.
In the future, apparently we’ll be featureless and wear latex suits with our genomes printed on them.
Now that sequencing costs have decreased, what are the other barriers to progress? A common theme that’s emerging from these WTFunding pieces it that being able to manage and analyze a lot of complex data turns out to be really hard…and bioinformatics is not different.
While it’s now far cheaper to generate genomic data, there’s still no guarantee that it’s accurate. Since gene annotation is still performed by humans, at least to some degree, there’s still plenty of room for human error — not to mention oftentimes the annotations are simply incomplete. You can play a “fun” game if you want and keep backing up the chain I walked you through earlier to spot the numerous chances for error to be incorporated, sequence assembly and sequencing itself being the most likely candidates.
You might have also caught while reading the “how it works” part that the algorithms for various processes require a lot of computational resources. Obviously the past decade has seen enormous strides in the availability of computing power, but it’s still reasonably time and cost prohibitive given the sheer size of genomic data. Someone else has already done the math on the size of the human genome in digital terms, and it’s roughly 200GB of raw sequencing data because, as explained before, you need a bunch of “reads” of the sequence.
On the data management side, a big question is how do you make genomic data easily accessible and combinable with other types of health data (such as patient outcomes), since that’s a key to unlocking a larger addressable market. There are startups addressing precisely this issue, in figuring out how to sensible combine different data types and link them up in an intuitive manner, but it’s safe to say that data harmony hasn’t yet been reached industry-wide.
The analysis portion specifically presents two big challenges. First, in a fragmented market for tools and algorithms and other analysis methods, how do you figure out what to use? Second, how do you know you’re not just seeing patterns in data that aren’t actually there? Many analysis tools are written with a single application in mind, and from what I gather, they aren’t very well-written, either. So organizations that could potentially benefit from bioinformatics may not be willing to dip their toe in the water until a discernable market with user-friendly software emerges.
And on the genetic testing side, the main questions are: Who pays for the tests? Who advises individuals on their test results? How does the data easily get to health care providers? While I’ll get into the incentive problems within genetic testing later, part of the problem currently is that genetic tests are still somewhat expensive for individuals. Will the government? With health insurance providers? It presents a bit of a chicken and egg problem, given patients need to be genetically tested in order to gain benefits from tailored pharmaceuticals or personalized medicine, but neither of those benefits are yet in a mature enough stage to incentivize people taking the tests.
Who cares?
 Pillz on ya billz
Pillz on ya billz
VCs love numbers, so I’m going to present the big, high-level ones first:
- Global pharmaceutical industry = ~$1 trillion annually
- U.S. pharma industry = ~$400 billion annually
- Global healthcare industry = ~$3 trillion annually
So combined, looking at a $4 trillion global market, which should assuage any VC’s fears of insufficient market size since grabbing 1% of the market gets you $40 billion in revenue. For reference, Pfizer has just about 5% of pharma market share and has a market capitalization of just over $200 billion.
But, the clever VC says, the initial markets will just be those with the highest pain points, not the entire industry! Those top therapy classes would be oncology (cancer), diabetes and mental health. So here are more numbers:
- Cancer drug spending in the U.S. = ~$42 billion annually (in 2014)
- Direct medical costs of diagnosed diabetes in the U.S. = ~$176 billion annually (in 2013)
- Direct cost of mental illness in the U.S. = ~$147 billion annually (in 2009)
VCs should still be quite happy, since combined (and given inflation), this is likely nearing a $400 billion market, and just in the U.S. alone. With the 1% market share goal, you’re still looking at $4 billion in revenue today.
Obviously, it isn’t just VCs who care about the big, shiny numbers. The aforementioned pharma companies likely get cartoon-dollar-sign-eyes contemplating the ability to charge more for genomic-data-driven drugs tackling these illnesses. Medical providers could also pad their pockets by leveraging personalized medicine to differentiate from the competition and thus create the ability to markup prices.
But on a less cynical note, there’s the real chance that genome-based personalized treatments won’t be solely a marketing shtick and actually result in significantly better outcomes for patients. So anyone suffering from an illness could potentially see benefits from personalized medicine. Further, there are many illnesses where the current understanding of what causes them can be summed up by ¯\_(ツ)_/¯, and bioinformatics could potentially lead to breakthroughs that would give hope of proper treatment and prevention to those afflicted.
What are the risks?
 And we all know how well Jurassic Park turned out
And we all know how well Jurassic Park turned out
There’s a pretty clear incentive problem on the pharmaceutical side. Pharma companies’ goal is to have more people buy their drugs. One way to incentivize more people to buy your drugs is by recommending certain drugs based on a person’s genetic profile. In a world in which genetic testing was performed by an altruistic, neutral third party, people might only be prescribed the drugs they need. In a world in which pharma companies have massive budgets, particularly for marketing, it isn’t difficult to imagine that they might “sponsor” genetic testing recommendations to ensure that their drugs are recommended.
As with any industry with a growing reliance on data-driven and increasingly automated approaches, there’s the potential for error as well. Relying too much on data models could mean that there is less caution than today towards prescribing treatments, which could result in disastrous “black swan” type events. Though, as I mentioned, it’s far more probable that healthcare providers will instead commit the error of seeing patterns that don’t actually exist, the implications of which could range from unnecessary and costly treatments to wrong and costly treatments that set back the patient’s progress, or even harm them.
Taking a look at another risk angle, the recent Theranos debacle is now a cautionary tale for medical startups. Many of the startups in bioinformatics are just in the software part of the space, so have less potential of scientific blundering like Theranos, since they’re instead helping with the data management, analysis and workflow portion.
But startups in the genetic testing area should definitely be heeding the lamentable tale of the “blood unicorn.” As I probably have made abundantly clear by this point, there’s room for error at nearly every step of the process, so there’s a non-trivial chance that end users might get inaccurate results that could have pernicious impacts on their health.
Let’s also not forget the cautionary tale of Jurassic Park, in which dinosaurs (which, spoiler alert, are extinct) are brought back to life through the power of DNA for the purposes of creating a theme park. Then everything goes to shit and people die. I’m pretty skeptical that anyone will be so silly as to bring back dinosaurs, but the general lesson of considering long-term implications is an important one. While the current research focus may be on disease prevention, there could be a future in which you can “enhance” yourself (or your offspring, like the “designer babies” I mentioned earlier) through genetic modification. It sounds really cool, but it could also be really, really disastrous and have far-reaching consequences not just health-wise, but societally (for anyone who has played Bioshock, think of ADAM).
On a geopolitical note, improvements in understanding the human genome also open up nefarious applications. At the “mildly evil” end of the evil scale, there’s genetic modification to win sporting events like the Olympics; at the “really evil” end of the evil scale, there’s creating highly targeted biological weapons to conduct warfare against a specific subpopulation, i.e. biological genocide. In fact, this year the U.S. Intelligence Community added gene editing to its “Worldwide Threat Assessment” report.
 Implanting needles in snakes’ mouths as fangs is clearly the most efficient bioweapon distribution mechanism…
Implanting needles in snakes’ mouths as fangs is clearly the most efficient bioweapon distribution mechanism…
Heretofore, the scale needed for biological weapons has limited it due to the rigorous research and development process that synthetic pathogens require. But thanks to improvements in sequencing technology and analysis, it’s far easier to sequence viruses, which means it’s potentially easier to create new viruses, too!
While it’s probably an exaggeration to say that most people could now make a bioweapon in their backyard, as the availability of genomic databases and computing power increases, and the cost of equipment continues to decrease, it will certainly be accessible to well-funded terrorist organizations (if not already).
I definitely don’t think it’s too tin-foil-y to suggest that most of the current world powers have synthetic bioweapons at their disposal already. However, it may be a bit more tin-foily to say that the population’s increasing reliance on antibiotics really doesn’t help proactively defend against some sort of biological attack.
Hopefully these bioweapons, like nuclear weapons, aren’t ever deployed in-the-wild and are instead more about deterrence. In any case, this sort of subject could make for thrilling TV.
What’s the current scene?
There are a number of startups scattered about the various areas within the bioinformatics chain, most of which seem to have early leaders emerging within them. As far as incumbents, there’s Illumina and Qiagen in the NGS part, CLC bio (owned by Qiagen) in bioinformatics analysis, Bina Technologies (owned by Roche) and Genomic Health in data management and most recently, Intellia in gene editing. I’ve segmented out the startups based on the different sub-areas below:
Data Management
- DNAnexus
- Genome Compiler
- Genospace
- MediSapiens
- SolveBio
- Benchling
- Omicia
Genomic Analysis
- Biomatters
- BioNano Genomics
- CancerIQ
- Maverix Biomics
- One Codex
- Onramp BioInformatics
- Spiral Genetics
- Syapse
- Tute Genomics
Genome Editing
- Caribou
- CRISPR Therapeutics
- Desktop Genetics
- Homology Medicines
Genetic Testing
- 23andMe
- Cofactor Genomics
- Color Genomics
- Counsyl
- NextGxDX
- Recombine
There isn’t one major investor that’s all over this space, though in the lead is definitely Google Ventures. Below is the list of the major institutional and corporate VCs that have played in this space in some form:
- Andreessen Horowitz
- Data Collective
- Felicis Ventures
- First Round
- Formation 8
- Google Ventures
- Johnson & Johnson
- Khosla Ventures
- Mohr Davidow Ventures
- New Enterprise Associates
- Novartis Venture Fund
- SV Angel
- WuXi Healthcare Ventures
- Y Combinator
Conclusion
 Bioinformatics startups will find a way
Bioinformatics startups will find a way
The implications of a more mature bioinformatics industry are pretty radical — from curing cancer to having vastly improved patient outcomes resulting from personalized medicine. I think it’s evident from the simplicity of some current solutions, such as basic data management and collaboration tools, that there’s a long way to go until this area becomes sufficiently sophisticated and automated.
While the error problem throughout the bioinformatics chain will certainly hinder adoption of end-applications, I think the truly massive dollar size amounts behind those applications will spur along innovation sooner rather than later. Perhaps even some of the washed out founders and engineers who are victims of the current market correction will apply their computer and data science skills towards this area — it seems a more easily accessible industry than others, with collaborative incumbents.
I think the biggest long-term opportunities are in what is some of the traditionally “boring” application infrastructure software — data management, collaboration, workflow management, etc. — both in pharmaceutical research and for healthcare providers. If I were a healthcare data platform like Flatiron Health or Health Catalyst, I might be looking to scoop up some of these companies early with the long-term vision of an integrated genomic and clinical data platform.
In any case, the companies that create this sort of middleware can use those data-driven underpinnings to very easily reach up into the sexier analytics and visualization plays. I honestly wouldn’t be shocked if some of these data management software startups become the SAP or Oracle of Bioinformatics.
In contrast, I think genetic testing will eventually become a commodity, which in my opinion doesn’t justify the level of risk that startups face with it today. Admittedly, it’s a crucial part of unlocking the potential of personalized medicine, but my gut feeling is that there will just end up being a Quest Diagnostics and Laboratory Corp. of genetic testing one day. For comparison, Quest has a market cap of ~$10 billion and Laboratory has one of ~$13 billion, while SAP has one of ~$95 billion and Oracle one of ~$163 billion (not to mention all of their competitors). So I think on the whole, the software bet is the smarter one given the risk / reward trade off.
Finally, I think we’re not at all yet prepared for the ethical challenges that will arise from bioinformatics applications…but that’s another long-read post for a different time.